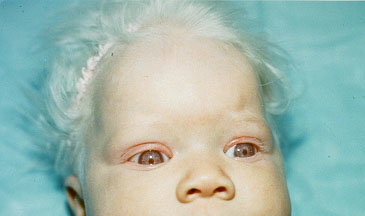
Turner syndrome is caused by a missing or incomplete X chromosome. People who have Turner syndrome develop as females. The genes affected are involved in growth and sexual development, which is why girls with the disorder are shorter than normal and have abnormal sexual characteristics.
Symptoms:
There are many different physical features associated with Turner syndrome. Not all girls have all symptoms, and in many cases the signs are hard to detect. Girls with Turner syndrome may have:
short stature (affects almost all girls with Turner, to different degrees)
failure of ovaries to develop (90-95% of girls)
webbed neck (25%) or short neck (40%)
abnormal fingernails and toenails (70%)
low hairline at neck (40%)
heart defect (30%)
kidney or urinary tract defect (30%)
hearing disorders (50-90%)
frequent ear infections in childhood (75%)
shortening of bones in the hands (35%)
lower jaw smaller than normal (60%)
drooping eyelids (ptosis), wandering eyes (strasbismus)
Girls and women with Turner syndrome have normal intelligence but often have learning problems that lead to difficulty with math and spatial relationships between objects.
Diagnosis:
If a physician suspects a girl may have Turner syndrome because she is not growing at a normal rate, and perhaps has one or more of the other signs of the syndrome, a chromosome analysis will be done. Finding the specific chromosome problem of the syndrome is the only definitive diagnosis.
Treatment:
There are two main medications given to girls with Turner syndrome. One is human growth hormone, used to increase the girl's growth rate and help her be taller. The other medication is estrogen, a female hormone, to replace the estrogen which would normally have been produced by the ovaries. Another female hormone, progesterone, is also given when the girl grows older, to help her have a normal monthly menstrual cycle.
Since a girl with Turner syndrome usually does not have ovaries, she cannot produce eggs and become pregnant when she grows up. However, some women with Turner syndrome can use in vitro fertilization to become pregnant, using donated eggs. Other women choose to adopt children in order to have a family.
source: click here